La difalia, diphallus o DP, es una rara anormalidad congénita cuya característica principal es que el hombre que la padece tiene dos penes. Se produce como consecuencia de algún fallo en la formación de los órganos genitales durante el desarrollo del feto, concretamente durante el primer mes de gestación.
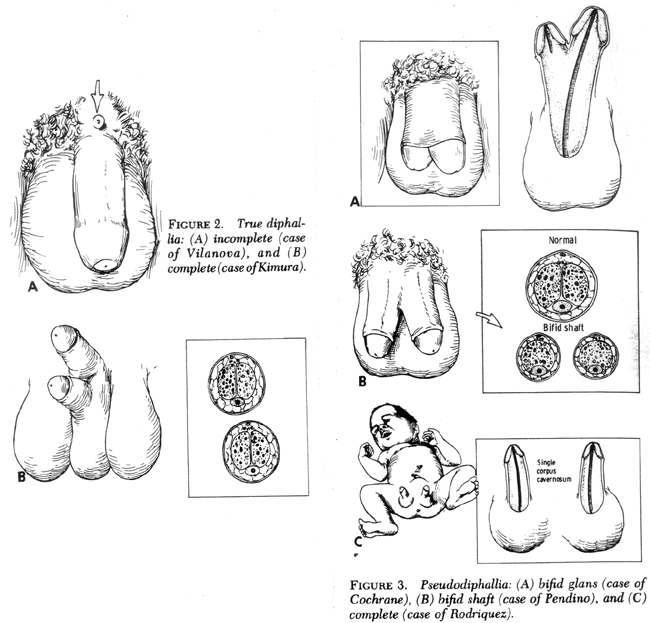
Existen distintas variaciones, siendo la completa la menos habitual. Esta consiste en presentar dos glandes independientes entre sí, dos cuerpos de pene y con perfecta funcionalidad cada uno de ellos.
Esta malformación suele presentarse junto a otro tipos de duplicidades, tales como la de riñones, la de intestinos, o en algunos casos asociados a la malformación producida por la conocida como espina bífida.
Hay tres casos que se pueden presentar:
- Uno es cuando existen dos glandes y dos troncos peneanos, pero la uretra la tienen en común, con el consiguiente trastorno para uno de ellos.
- Otro es la difalia verdadera, DP, que es cuando existen dos penes completos y, además, ambos poseen su propia uretra.
- Luego está la conocida como pseudodifalia, que es cuando uno de los penes se presenta vacío,es decir, que no es un propiamente un cuerpo cavernoso.
Según las estadísticas, esta anomalía la sufre uno de cada 5,5 millones de miembros del sexo masculino, con lo cual se confirma la inusual que es su aparición. El primer caso del que se tiene noticia es del año 1609 y, desde entonces, sólo se han contabilizado un centenar de casos más.
Como anécdota cabe destacar que uno de los últimos casos, y que además, ha contado con la divulgación de los medios de comunicación, es el de un hombre de 24 años, casado y de origen hindú, que ha pedido que a pesar de tener dos penes que funcionan correctamente, se le extirpe uno, pues desea tener una vida sexual placentera y, sobre todo, normal.

Según datos estadísticos 1 de cada 5,5 millones de estado unidenses tiene dos penes.